
—— Съвместно проучване на Zhejiang CDC, Macro & Micro-Test и China CDC, публикувано във Frontiers in Cellular and Infection Microbiology
Преглед на проучването
През май 2026 г. Frontiers in Cellular and Infection Microbiology (JCR Q1, IF ≈ 4.6) публикува статия, ръководена от Центъра за контрол и превенция на заболяванията в провинция Джъдзян (Zhejiang CDC), с екипа по биоинформатика от Beijing Macro & Micro-Test Bio-Tech Co., Ltd. и Националния институт за контрол и превенция на заразните заболявания (China CDC) като съавтори. Изследването е озаглавено:
„Идентификация и филогенетичен анализ на седем щама Brucella abortus в Джъдзян, Китай.“

Това проучване представлява първият систематичен, базиран на целия геном филогенетичен анализ на проследимостта на Brucella abortus (B. abortus) в провинция Джъдзян, Китай. Екипът анализира седем изолата, събрани от 2015 до 2025 г. (четири щама с човешки произход и три щама с говежди произход от Джинхуа, Цюйчжоу и Нингбо). Констатациите предоставят геномни доказателства за произхода и пътищата на предаване на този „северно доминиращ вид“ в нетипичен южен епидемичен регион в източен Китай.
Предистория и значение
Бруцелозата е зоонозно заболяване, причинено от бактерии от рода Brucella. Brucella abortus заразява предимно говеда, но може да причини заболяване и при хора. В Китай бруцелозата показва значителни географски вариации: най-високата честота се наблюдава в северните провинции (напр. Вътрешна Монголия, Шанси, Хейлундзян). За разлика от това, южните провинции, включително Джъдзян, исторически са били доминирани от Brucella melitensis, с много малко докладвани случаи на B. abortus. Това регионално неравенство прави генетичната характеристика и проследяването на източниците на B. abortus в Джъдзян ключов приоритет за общественото здраве.
Методи и ключови открития
Изследователският екип възприе многостранна стратегия, съчетаваща молекулярна биология и биоинформатика:
1.Идентифициране на патогени и основно типизиране
PCR на гена BCSP-31 и AMOS-PCR потвърдиха, че и седемте изолата са B. abortus.
Многолокусното секвенционно типизиране (MLST), базирано на девет гена за поддържане на гени, разкри, че всички изолати принадлежат към секвенционен тип ST2, което показва висока генетична хомогенност сред циркулиращите щамове на B. abortus в Джъдзян.
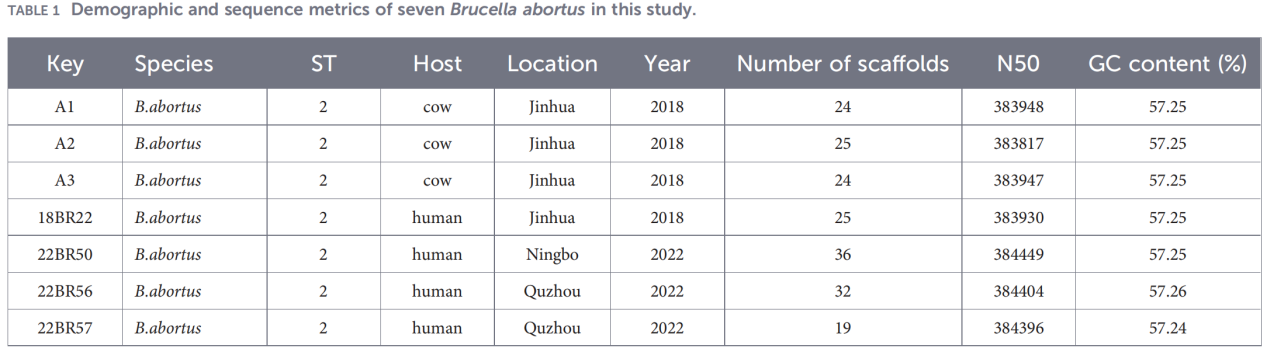
2.Характеризиране на целия геном
Секвенирането на целия геном беше извършено на платформата Illumina NovaSeq. Анализът на средната нуклеотидна идентичност (ANI) показа, че изолатите от Zhejiang споделят до 99,99% сходство с референтния щам B. abortus 544.
Пан-геномният анализ разкри силно консервирана популация: идентифицирани са 3084 основни гена, заедно само с 10 гена на черупката, и не са открити меки основни или облачни гени.
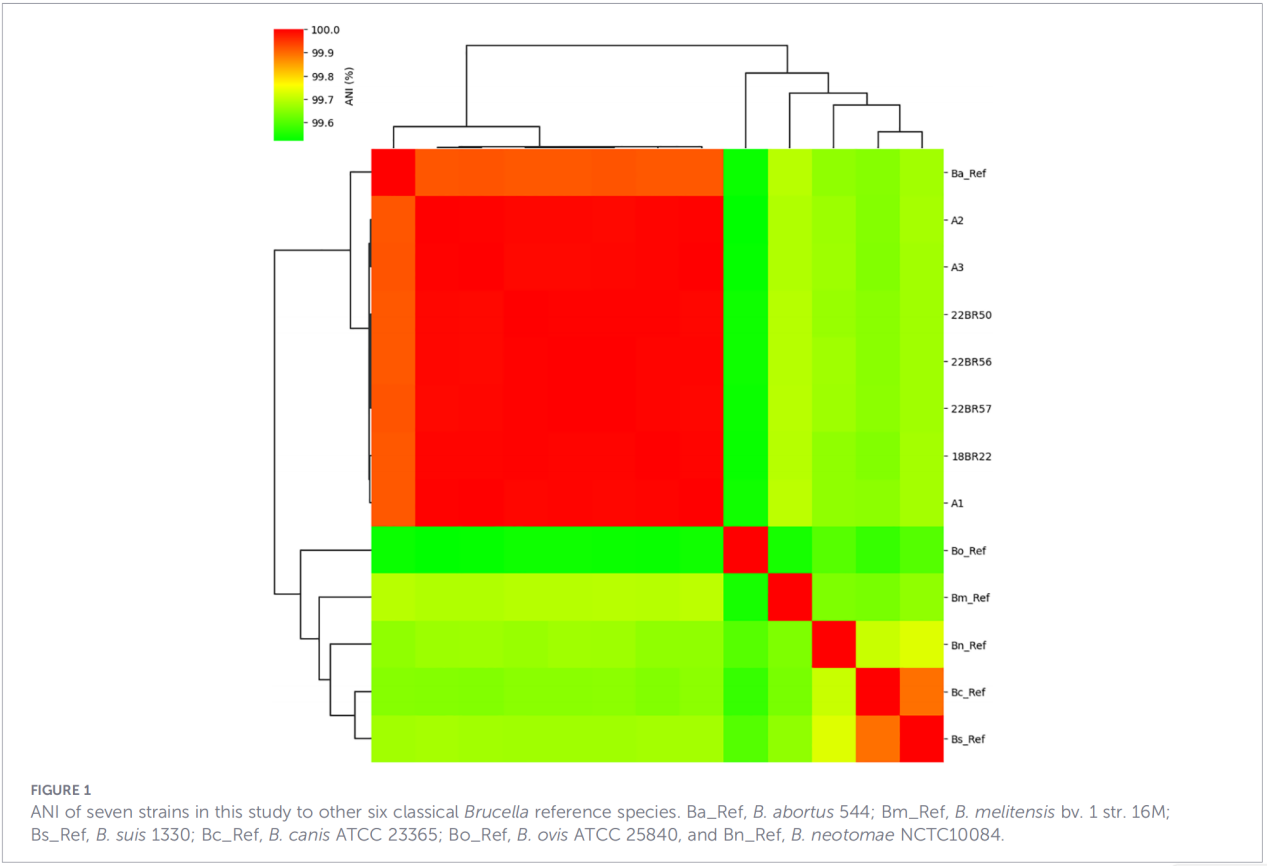
3.Профили на гени за вирулентност и антимикробна резистентност
Предсказани са общо 68 фактора, свързани с вирулентността, обхващащи класически пътища като биосинтеза на LPS, секреторната система на T4SS и двукомпонентната регулаторна система BvrR-BvrS. Забележително е, че всички изолати не са имали адхезиновите гени bmaA и btaF. Анализът на гените за резистентност е открил само гена mprF в базата данни CARD, без да са идентифицирани други детерминанти на резистентност.
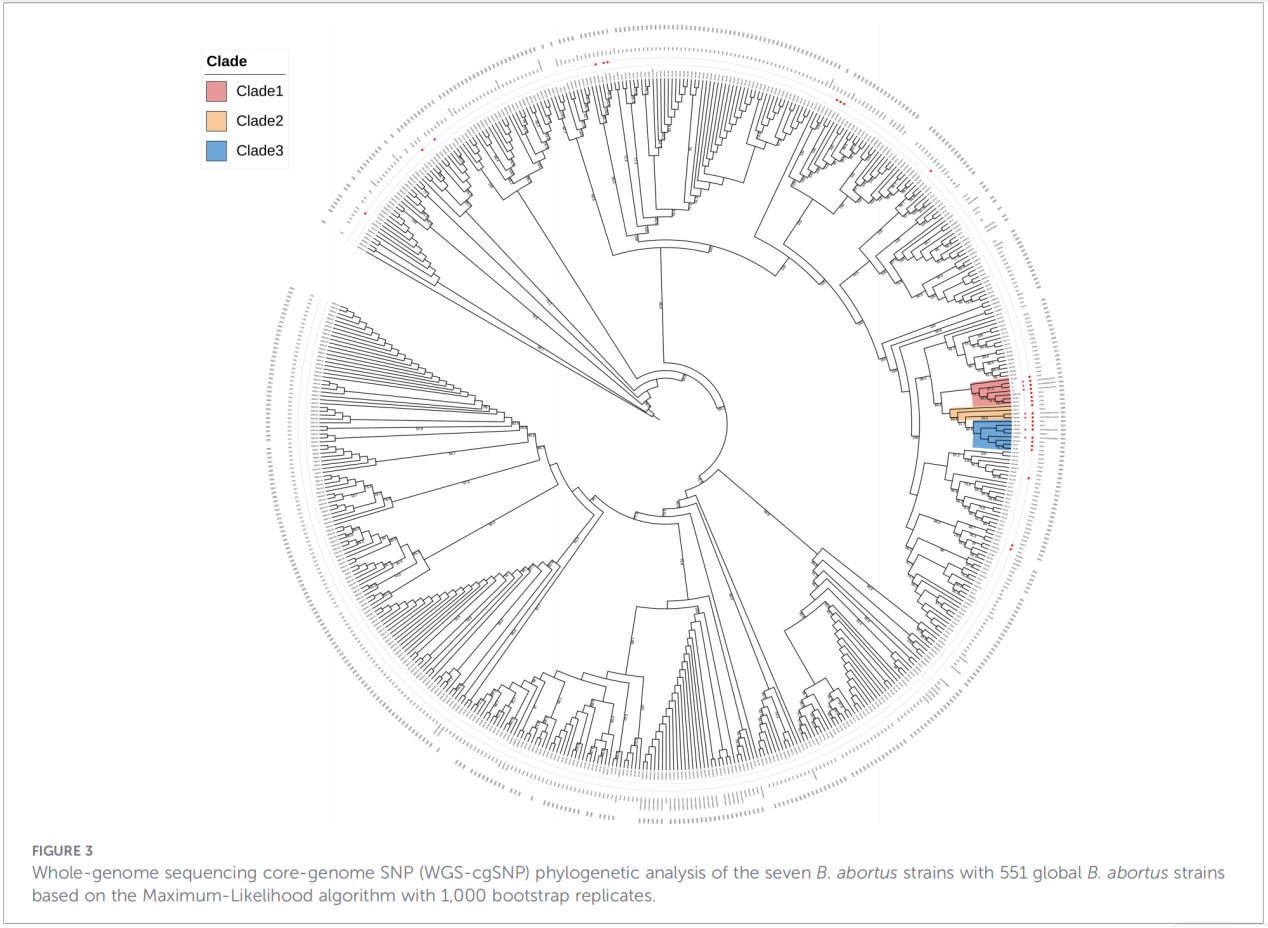
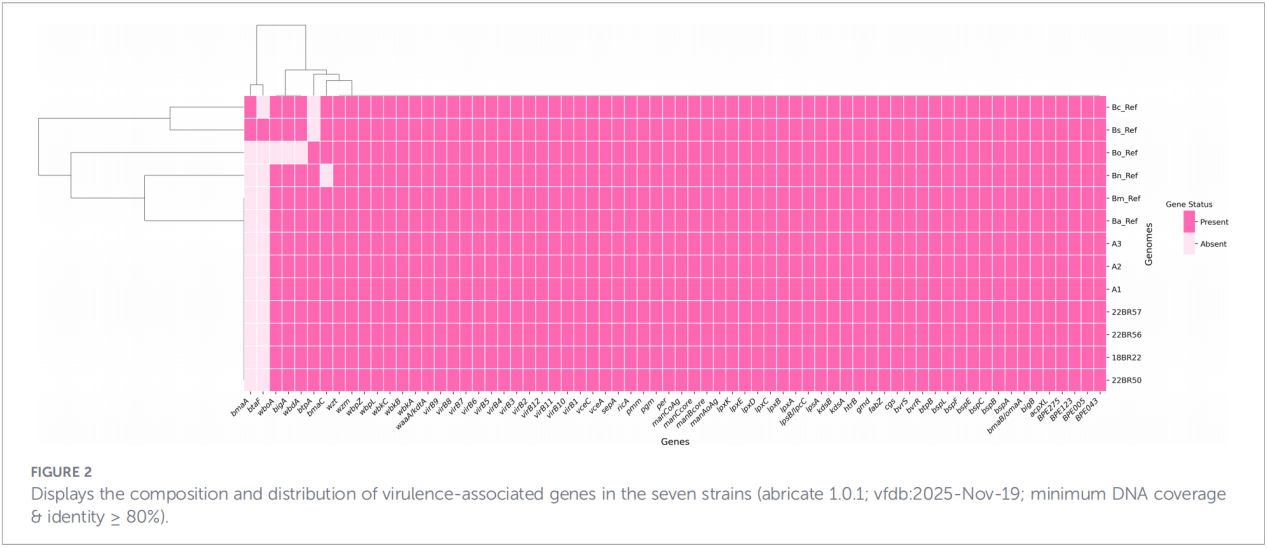 4. Филогенетична реконструкция и проследяване на предаването
4. Филогенетична реконструкция и проследяване на предаването
Анализът на еднонуклеотидния полиморфизъм в ядрото на генома (cgSNP) постави изолатите от Джъдзян на специфична позиция в глобалното филогенетично дърво. Резултатите показаха, че щамовете от Джъдзян образуват монофилетична група заедно със щамове от Русия, Монголия и няколко северни китайски провинции (Нинся, Хейлундзян, Вътрешна Монголия, Хъбей, Гансу, Пекин). Тази група се разделя допълнително на три отделни подклада (клад 1–3), което предполага множество независими събития на въвеждане.
Заключения и последици
Това проучване предоставя първия високопрецизен геномен набор от данни за B. abortus в провинция Джъдзян и води до няколко ключови заключения:
- Клеar генетичен фон– Щамовете на B. abortus, циркулиращи в Джъдзян, принадлежат към ST2, геномно са силно консервирани и представляват типична линия на бруцелоза по говедата.
2. Евина междурегионалното предаване– Филогенетичният анализ не подкрепя съществуването на независим ендемичен род в Джъдзян. Вместо това, данните категорично показват, че тези щамове произхождат от Северен Китай и може да споделят общ еволюционен произход със щамове от Русия и Монголия. Наличието на три подклада предполага множество отделни събития на въвеждане.
3. Последици за общественото здраве– Констатациите подчертават стойността на геномното наблюдение за бруцелоза дори в традиционно неендемични региони като Джъдзян. Въпреки че настоящият брой на случаите е нисък, инструменти с висока резолюция като cgSNP могат ефективно да проследят източника на внесени огнища и да предоставят научни доказателства за прекъсване на веригите на предаване, свързани с междупровинциалния транспорт на добитък.
Тази работа не само запълва празнина в изследванията в провинция Джъдзян, но и предоставя нови базови данни за наблюдение на патогените и оценка на риска от бруцелоза в района на делтата на река Яндзъ.
Информация за документа:
Yang, Y., Shi, X., Chen, J., Wang, L., Wu, Z., Yao, W., … & Wu, B. (2026). Идентификация и филогенетичен анализ на седем щама Brucella abortus в Джъдзян, Китай. Frontiers in Cellular and Infection Microbiology, 16, 1758965.
Време на публикуване: 10 юни 2026 г.